
Boundary Conditions in Space and Time
How do we calculate distances and energies in periodic boundary conditions? In the minimum image convention we use the nearest distance among all the images. This implies that we must smoothly truncate the potential at L/2. Otherwise energy will not be conserved as particles switch from one image to another. See the pseudocode for force calculation in periodic boundary conditions. We will discuss next what happens to long range potentials in PBC.
It is possible to use any regular space filling lattice (Bravais) for boundary conditions such as the truncated octahedron since it is more spherical. This is also necessary when simulating non-cubic crystal structures.
It has been found that one hundred atoms in PBC can behave like an infinite system in many respects. The corrections are order (1/N) and the coefficient can be small. One cannot calculate long distance behavior (rL/2) or at large times (the traversal time for a sound wave.) Angular momentum is no longer conserved. Also PBC can influence the stability of phases. For example, PBC favor cubic lattice structures that fit well into the box.
Neighbor Tables for Short Ranged Potentials
We have already discussed how to cutoff short-ranged potentials at the box edge, necessitated by the need to have a continuous force for molecular dynamics. But what happens as the number of particles grows? Clearly the code fragment the operation count and hence the CPU scales as N2. This is its complexity.
But as the system gets bigger we do not have to keep increasing the cutoff radius. Once the cutoff radius gets outside the second neighbor distance, or if V(rc) < kBT we can fix rc. Then use of a more efficient scheme can reduce the complexity to N.
Suppose we have a potential between pairs of particles in vacuo which goes like v(r). There are really these two distinct issues which sometimes get confused:
Vimage (r) = S v(r+image) - background.
The background is put in to make the sum converge. If one sums spherical shells, the series is conditionally convergent. However one has a choice about surface charges. A particle interacting with another particle will see an infinite array of the other particle. One has to decide if this is physically correct. Inhomogeneous systems like a single charged impurity are problematical. One is essentially computing the interaction at a low but finite concentration. The Ewald method has the great advantage that it respects the Poisson equation. This implies that the long-wavelength limits are correct.
The second question is a little more subltle. Essentially the idea is
What tests can you make for your Ewald code? You can put the charges on a perfect lattice and make sure that you get the known Madelung constants.
In the last 10 years methods have been developed to do this computation with complexity N. The older methods are called particle-cell methods. More recently has been developed the fast multipole method, see R. Hockney and J. Eastwood, Computer Simulation using Particles (McGraw-Hill, NY, 1981). There has also been Fast Fourier Poisson Method (e.g., D. York and W Yang, J. Chem Phys. 101, 3298 (1994)) which is O(N ln N). For a good overview of these and other methods, see Toukmaji and Board, Jr. at Duke.
Static Correlations and Order Parameters
Today, I will discuss some of the most important information that comes from simulations, the static and dynamic correlation functions. You should develop a habit of looking at these quantities routinely since they tell you basic information about what the particles are doing, namely where they are and how they arrange themselves.
Most of the information from this talk is on the formula page pdf.
The types of static correlations can generally be divided into
You find it by histogramming, that is subdividing the total volume into a set of subvolumes.
How to histogram:
Three dimensional histograms are a problem because, the number of hits/bin is low, they are hard to visualize, they take alot of memory. You might want to use symmetry or projections to make it into a 1 or 2 dimensional density.
It is also good to examine the density in Fourier space. In periodic boundary conditions the allowed K-vectors are on a grid. The Fourier density can be used to either: 1) smooth out the real space part by setting to zero the FT density larger than some value and F transforming back. (Disadvantage: the resulting density might go negative.) 2) look for periodic structures.
The Fourier transform of g(r) is called the static structure function Sk. It is important to calculate because it can be compared directly with experiment, either neutron or X-ray scattering can measure it. (there are very expensive instruments at Argonne, Grenoble, Brookhaven that are dedicated to measuring Sk and its time dependent generalization. If the agreement is good it gives a lot of confidence concerning the potential model. But don't be carried away; agreement to 5% is typical if the potential is at all reasonable.
The first peak of Sk is around 6 density 1/3. Normalization is such that at large k: Sk=1. It can be computed in two different ways, either as a FT of g(r) or the direct way. The direct way is slower but better at low k-values or a reciprocal lattice vectors of a possible solid structure because there are not the systematic errors of ignoring anisotropic correlations and histogram effects. Also g(r) must be extended to large r to do the FT.
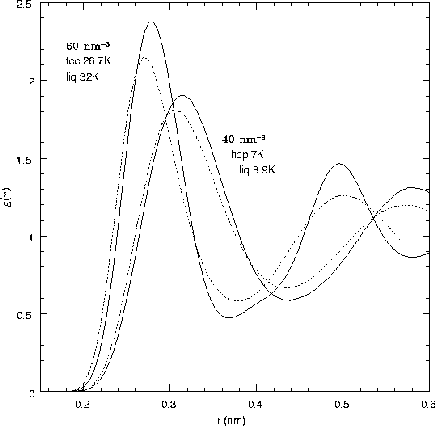
One should use the peak of Sk to decide is a system is solid. For a liquid Sk is smooth and order 1 everywhere. A solid shows a delta functions with peak high a fraction of the number of particles at the reciprocal lattice of the solid. Solid structure shows up in g(r) in a more subtle way: instead of a nice damped oscillations, there are bumps at the nearest neighbor, next-nearest neighbor, etc. (Where they are depends on the type of lattice). See the figures for illustrations of g(r) in liquid helium.
The structure factor also signals separation in a two phase region. An example is the formation of droplets such as occurs when the number of particles corresponds to neither a stable liquid of gas, but a mixture of liquid and gas. In that case the structure function will diverge at low wave vector. The value at zero (S0) equals N by definition (in the canonical ensemble) but this can be different than limit {k Þ 0} Sk which is proportional to the compressibility of the system.
Order parameters
A key concept from the theory of phase transitions is the idea of an "order parameter" which characterizes a phase. For the moment we will just explain how this works for the two most common phase transitions: liquid-gas and liquid-solid.
The liquid-gas transition: the order parameter is the density. a liquid and gas are identical except a liquid is higher density. At the critical point the difference disappears. There is a first order phase transition between liquid and solid so it is quite possible to do a simulation in the two phase region. What will happen? Liquid droplets will form assuming 1) the surface tension allows it an 2) the simulation is long enough. When this happens Sk will have a characteristic behavior.
The liquid-solid transition : A solid is characterized by a periodic density. For example the Fourier transform of the density will be very large for certain wavevectors (the reciprocal lattice vectors) CLAMPS allows you to input a particular set of vectors to monitor melting and freezing. (keyword READ_K file )
Correlation Functions for Dynamical Response
See the pdf formula page. Readings A & T pgs. 58-64, 185-191
Now consider the calculation of dynamical properties and the response to perturbation. For static properties it would probably be more convenient to do Monte Carlo methods since they are more robust. Dynamics is the reason that we do MD.
The simplest dynamical properties are the linear response to an external perturbation. Most macroscopic measurements are in the linear regime because macroscopic perturbations are very small on the scale of microscopic forces.
Onsager's made a well known conjecture (1935), called the Regression Hypothesis. (also known as the fluctuation-dissipation theorem.) It states that the linear response of the system to a time-varying perturbation is the same as fluctuations that naturally occur in statistical equilibrium. So there are 2 basic ways to calculate response: either wait for the system to fluctuate by itself or apply a perturbation and see what happens. (the first is often referred to as the Kubo method.)
H = H0 + c A
where H0 is the unperturbed Hamiltonian, A is a perturbation and 'c' is the coupling constant. As shown on the formula page one can write the free energy as a power series in 'c'. The linear response is simply the expectation of A in the unperturbed system. The second order term is the fluctuation about the mean. In fact if we neglect all terms above the first order we always get an upper bound to the free energy. Finally to calculate the free energy difference between the system at c=0 and c=1 we just need to integrate the average A over ensembles for various values of c between 0 and 1.
We can also find the response of some property (B) to the perturbation; it equals the correlation between A and B. For example one can find the effect of cutting off the potential. Generally one expects that if the neglected potential is smooth it is pretty much independent of the phase space point R, and hence terms higher order than first can be neglected. A second example is to apply a sinusoidal potential and look at the response of the density. It is simply S k, the structure factor. That is essentially how neutrons and X-rays measure the structure factor.
The transformation assumes that one is using the unwound coordinates; the positions of the atoms such that:
r(t+h) = r(t) + h v(t) + ...
Don't use periodic boundary conditions to put particles back in the central cell.
One of the key discoveries in the early days of computer simulation was the long-time tails on the v-v function. by Alder and Wainwright. The v-v correlation functions decay algebraically in time. (as t-d/2) where d is the spatial dimension.) This implies that diffusion may not really exist in 2D. This is not Markovian (random) behavior which would decay exponentially in time. The momentum of a fast moving particle gets stored in the nearby liquid as a vortex and helps to keep it moving in the same direction for a very long time. (citation)
The dynamic structure factor is a key quantity (also known as the
density-density response function). If you push the system at time and place
(r,t) how does it respond at (r',t')? This is what neutron
scattering does. It is not hard to see that sound waves of various kinds will
show up as prominent features of Sk (w). This is a good way to
see them. The book The Theory of Simple Liquids by Hansen and MacDonald
contains a thorough treatment of the theoretical properties of this function.